Doença de von Willebrand: diagnóstico e cuidados

Erick Adolf von Willebrand, como muitos outros descobridores de condições médicas, deu seu nome à doença de von Willebrand (DvW). Sua jornada de descoberta começou com uma família, mais precisamente, com uma menina de 5 anos que sofria de um grave quadro hemorrágico.

Infelizmente, a garotinha faleceu aos 13 anos durante seu quarto período menstrual. Erick observou alguns sinais que o levaram a concluir que a garota estava exibindo sintomas de uma doença hereditária. Outros parentes, incluindo os pais, também apresentavam sintomas hemorrágicos, embora menos intensos. Além disso, a menina já havia perdido quatro irmãs, todas com menos de 4 anos de idade, todas elas exibindo distúrbios hemorrágicos.
A compreensão de von Willebrand
Erick estabeleceu uma base sólida para seus estudos ao concluir que a condição era hereditária e autossômica. Testes como o tempo de coagulação (normal) e o tempo de sangramento (prolongado) foram úteis na classificação da doença.
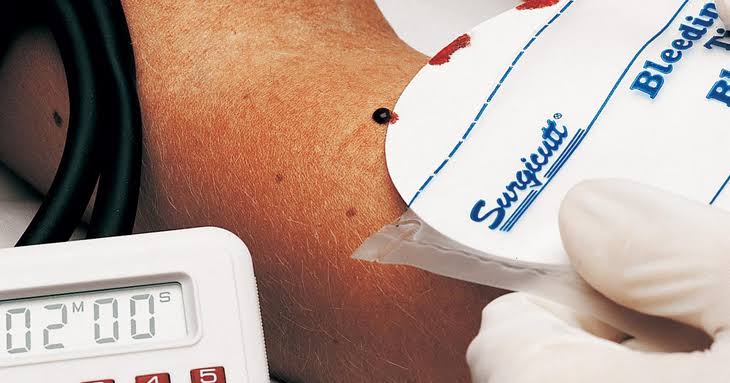
O tempo de coagulação antecedeu o TTPA (Tempo de Tromboplastina Parcialmente Ativado), que avaliou a coagulação intrínseca. Na deficiência do fator de von Willebrand (FvW), as plaquetas não aderiram adequadamente ao vaso, causando sangramentos, o que indicava problemas no vaso ou na interação plaqueta-vaso, mas não havia uma definição precisa na época.
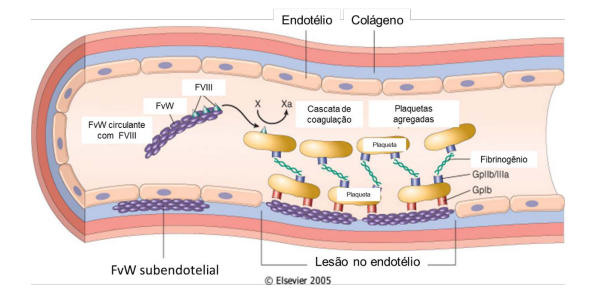
O que é o FvW?
As células endoteliais e megacariócitos produzem uma proteína polimérica conhecida como Fator de von Willebrand (FvW). Essa proteína desempenha duas funções principais: 1 – Age como uma ponte entre o colágeno exposto em um vaso lesionado e as plaquetas, o que facilita a formação eficaz de um tampão plaquetário; 2 – Garante a disponibilidade adequada do Fator VIII (FVIII). Portanto, a deficiência de FvW também pode resultar em deficiência de FVIII, um aspecto de crucial importância para a coagulação.
O mecanismo da patologia
A maioria dos casos de doença de von Willebrand resulta de mutações no cromossomo 12, que contém informações para o Fator de von Willebrand (FvW). A transmissão dessa condição ocorre principalmente por herança autossômica dominante, embora também existam tipos relacionados a outras heranças genéticas.
Essas mutações causam a troca de um aminoácido devido a uma única mutação no DNA, tornando a doença altamente heterogênea, com várias possibilidades de combinações genéticas.
A doença de von Willebrand afeta cerca de 1% da população, e 1% desses indivíduos manifesta sintomas leves a moderados. A diversidade de mutações dificulta a previsão da diminuição dos níveis de Fator VIII e, consequentemente, o prolongamento do Tempo de Tromboplastina Parcialmente Ativado (TTPA).
A hemostasia, influenciada por diversos fatores, pode causar sinais de sangramento mesmo em pacientes com níveis normais de Fator VIII. Os médicos realizam vários testes para aprimorar o diagnóstico. Além do TTPA, que é um teste de triagem, existem outros testes usados para identificar a doença de von Willebrand e diferenciar seus tipos e subtipos.
- Atividade do fator VIII coagulante (fator VIII:C);
- Antígeno do fator de von Willebrand (FvW:Ag);
- Atividade do cofator de ristocetina (FvW:RCo);
- Ligação do FvW ao colágeno;
- Aglutinação plaquetária com ristocetina (RIPA);
- Multímeros do fator de von Willebrand.
Achados clínicos
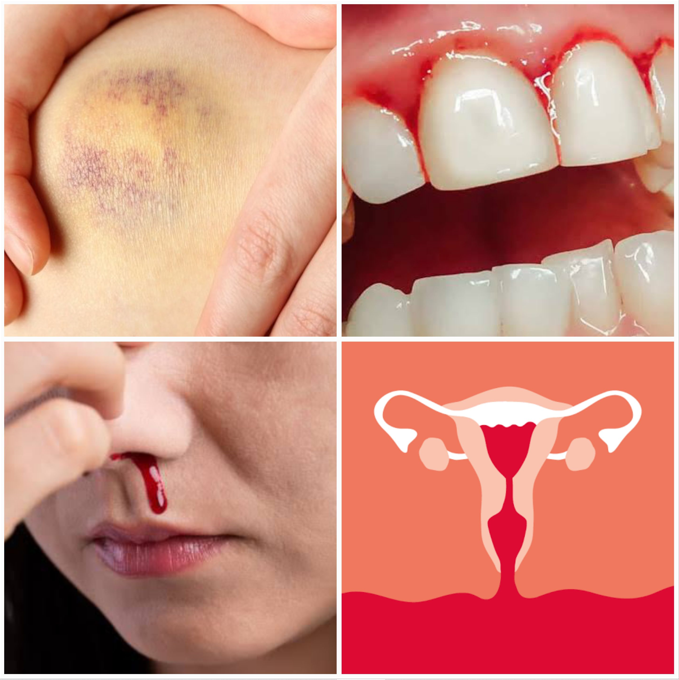
Os achados clínicos da DvW variam dependendo da gravidade da doença, dos tipos e subtipos, resultando em diferentes manifestações. Em casos moderados a graves, os principais sintomas incluem equimoses, sangramento gengival, sangramento nasal e, no caso das mulheres, fluxo menstrual anormal. Essa doença é de origem genética, e os sintomas geralmente se manifestam na infância ou adolescência, mas podem ocorrer em qualquer fase da vida do portador.
Doença de von Willebrand adquirida
A doença de von Willebrand adquirida pode ocorrer espontaneamente ou estar ligada a doenças que reduzem os níveis de Fator de von Willebrand (FvW) devido a diversos mecanismos. Isso inclui a presença de anticorpos contra o FvW, um aumento na proteólise (quebra) do FvW, a ligação do FvW a células (geralmente células tumorais) ou uma diminuição na síntese do FvW, como ocorre em desequilíbrios hormonais.
Referências
- HOFFBRAND, A. V.; MOSS, P. A. H. Fundamentos em Hematologia. Artmed. 7 ed. 2018;
- SBAC. Tratado de Análises Clínicas. Atheneu. 1 ed. 798p. 2018. ZAGO, M. A.; FALCÃO, R. P.; PASQUINI, R. Tratado de hematologia. Atheneu. 899p. 2013;
- ZAGO, M. A.; FALCÃO, R. P.; PASQUINI, R. Tratado de hematologia. Atheneu. 899p. 2013
- Ministério da Saúde. Manual de Diagnóstico e Tratamento da Doença de von Willebrand. Disponível em: https://bvsms.saude.gov.br/bvs/publicacoes/manual_tratamento_willebrand.pdf. Acesso em: 12 de setembro de 2023.




0 Comentários