SÍNDROME HEMOFAGOCÍTICA
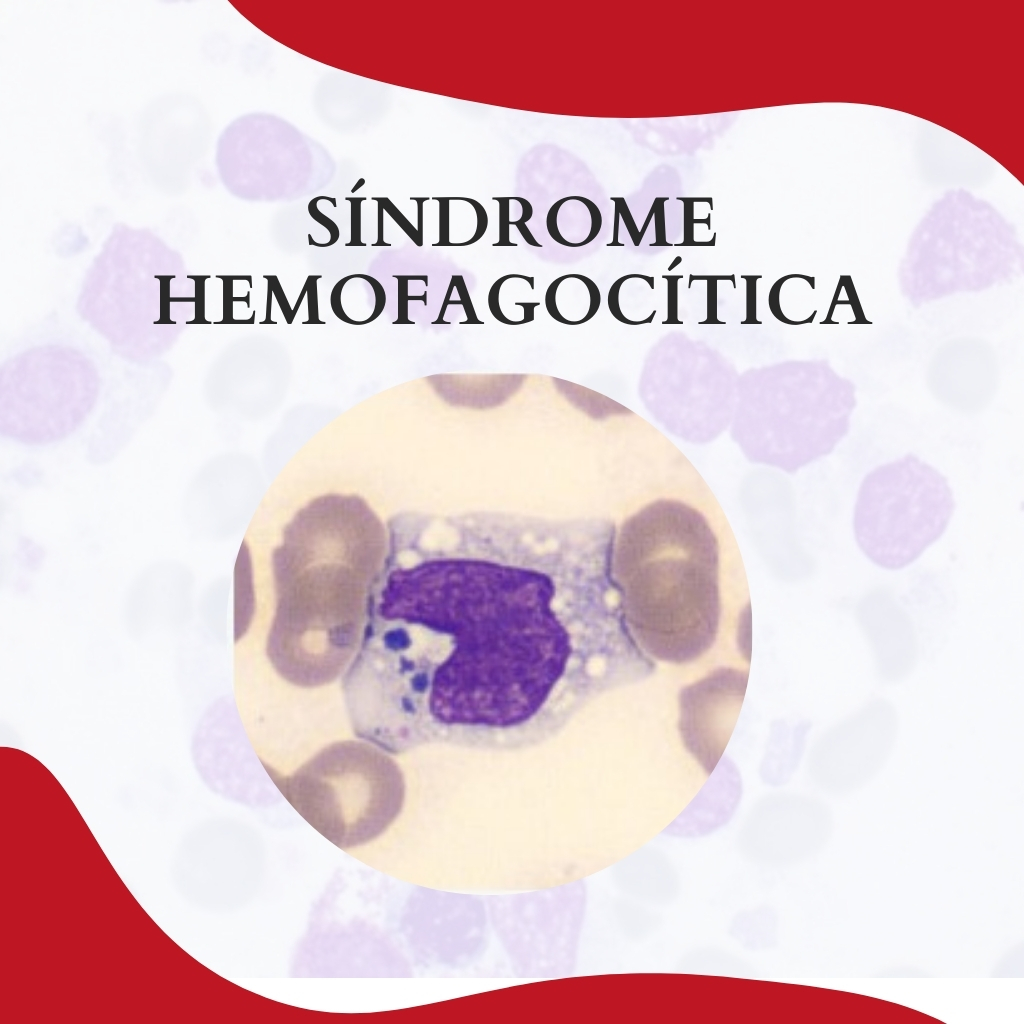
A síndrome hemofagocítica (SH) é uma condição séria desencadeada por uma hiperativação na resposta imunológica que desencadeiam a ativação generalizada de macrófagos levando a um processo inflamatório agudo grave e rapidamente progressivo.
Há duas formas dessa síndrome: A primária, causada por mutações genéticas que afetam a função das células TCD8 e Natural Killers, ocorrendo principalmente na infância (com maior incidência em crianças menores de dois anos – 80% dos casos), conhecida como Linfohistiocitose Eritrofagocítica Familiar. Esta forma geralmente tem um histórico familiar positivo ou envolve consanguinidade e é caracterizada por mutações recessivas autossômicas no gene da perforina-PRF1 (40%). A perforina é uma proteína presente em linfócitos, macrófagos e outras células precursoras da medula óssea, que desempenha um papel nos processos citotóxicos, na ativação contínua de linfócitos e no aumento das citocinas, levando à ativação de macrófagos.
Por outro lado, a forma secundária está associada a processos infecciosos em cerca de 50% dos casos, seguidos por casos neoplásicos, reumatológicos e autoimunes que podem afetar adultos de todas as idades. Essa forma também é chamada de síndrome de ativação macrofágica. Na SH secundária, as infecções agudas podem ser tanto primárias em indivíduos saudáveis quanto desencadeadoras em pacientes com condições autoimunes ou neoplásicas pré-existentes. Entre os patógenos envolvidos, as infecções virais são as mais comuns, especialmente aquelas causadas pelos vírus Epstein-Barr (EBV) e citomegalovírus (CMV).
Devido ao quadro inflamatório disseminado, as principais alterações laboratoriais incluem o aumento de ferritina, disfunção hepática, com altos níveis de lactato desidrogenase, AST, ALT, TP e bilirrubina total. As enzimas hepáticas podem ter um incremento 3 vezes maior que valor de referência. Outras alterações podem incluir a hipertrigliceridemia, que ocorre devido à inibição da lipoproteína lipase pelo TNF-alfa, elevação do D-dímero, causada pela coagulação intravascular disseminada.
O nível de Gama Glutamil Transferase (GGT) também se encontra aumentado, em função da infiltração do trato biliar pelos linfócitos e macrófagos. No hemograma ocorre leucocitose, e é possível observar em lâmina uma monocitose com presença de grandes monócitos ativados:
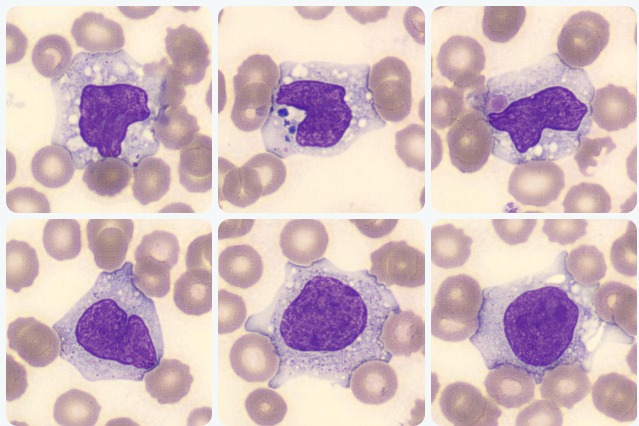
Por fim, a síndrome hemofagocítica é uma condição grave que requer uma abordagem multidisciplinar para diagnóstico e tratamento. Embora seja uma condição rara, é importante considerá-la em pacientes com sintomas sugestivos, especialmente em indivíduos com doenças subjacentes predisponentes.
O próximo passo de todo analista que deseja ter mais segurança na bancada
Todo analista que busca se destacar e se tornar um profissional mais atualizado, capacitado e qualificado para o mercado de trabalho precisa considerar uma pós-graduação.
Um profissional com especialização é valorizado na área laboratorial; esse é um fato inegável.
Unimos o útil ao agradável ao desenvolver uma pós-graduação em Hematologia Laboratorial e Clínica.
Para aqueles que procuram a comodidade de uma pós-graduação 100% online e ao vivo, sem abrir mão da excelência no ensino, temos a solução ideal.
Nossa metodologia combina teoria e prática da rotina laboratorial, garantindo um aprendizado efetivo.
Contamos com um corpo docente altamente qualificado, com os melhores professores do Brasil, referências em suas áreas de atuação.
No Instituto Nacional de Medicina Laboratorial, temos apenas um objetivo: mais do que ensinar, vamos tornar VOCÊ uma referência.
Toque no botão abaixo e conheça a pós-graduação em Hematologia Laboratorial e Clínica.
QUERO CONHECER TODOS OS DETALHES DA PÓS-GRADUAÇÃO
Referências
VIEIRA, Karoline Araujo; SOUZA, Gustavo Henrique Gandolfo. Síndrome hemofagocítica: um relato de caso. Brazilian Journal of Development, v. 9, n. 1, p. 248-255, 2023.
JÚNIOR, Ivo Ronchi et al. Síndrome hemofagocítica. Relacionado ao caso. Revista da Sociedade Brasileira de, v. 5, pág. 382-8, 2011.
JORDAN, Michael B. et al. How I treat hemophagocytic lymphohistiocytosis. Blood, The Journal of the American Society of Hematology, v. 118, n. 15, p. 4041-4052, 2011.



0 Comentários