A talassemia Beta é a forma mais comum de talassemia no Brasil, caracterizada por uma diminuição ou ausência na síntese de cadeias beta de globina. O gene defeituoso apresenta geralmente mutações pontuais (troca, acréscimo ou deleção de um ou dois nucleotídeos) envolvendo a regulação ou expressão do gene produtor da cadeia beta.
O gene da cadeia β de globina está presente no cromossomo 11, juntamente com os genes das outras cadeias globínicas delta e gama. Existem ao menos 200 tipos de mutações envolvidas com as beta-talassemias, embora cerca de 20% deles respondem por 80% dos casos mundiais. Mesmo com as diversas mutações, são identificados dois tipos de gene betalatassêmicos:
- Um gene totalmente incapaz de produzir cadeia beta (β0)
- Um gene que produz uma pequena quantidade de cadeia beta, mas inferior da normal (β+)
Assim, os genótipos possíveis de encontrar são:
● β/β (Pessoa normal)
● β0/β0 e β+/β+ (Homozigotos)
● β0/β+ (duplo heterozigoto)
● β0/β e β+/β (heterozigotos)
Vejamos o que acontece…
Em um extremo, os homozigotos para o gene β0 (genótipo β0/β0) não Produzem absolutamente nenhuma cadeia beta, e os duplos heterozigotos (genótipo β0/β+) produzem pouquíssima cadeia beta. Em ambos os casos o paciente desenvolve a beta-talassemia maior (anemia de Cooley), um quadro de extrema gravidade e letalidade, dependente de hipertransfusão para a sobrevivência.
No extremo oposto, os heterozigotos para o gene β0 ou β+ (genótipo β0/βou β+/β) apresentam a chamada beta-talassemia menor ou “traço talassêmico”. Um gene produz cadeia beta normalmente, enquanto o outro não produz ou produz menos. Estes indivíduos são totalmente assintomáticos, embora tenham típicas alterações nos parâmetros do hemograma. Com anemia microcítica e hipocrômica, além da presença de codócitos e pontilhado basófilo. Os homozigotos para o gene β (genótipo β+/β+) costumam dar origem à beta-talassemia intermédia, um quadro moderadamente grave de talassemia, mas não dependente da hipertransfusão. No geral, a gravidade da anemia e do quadro clínico é dependente da quantidade total de cadeia beta produzida pelos dois alelos.
A Diminuição das cadeias beta nas Células Eritroides
Existe duas consequências prováveis a diminuição das cadeias beta nas hemácias, a primeira é que ocorre uma diminuição na síntese de hemoglobina, promovendo microcitose, hipocromia e anemia; a segunda é que sobram cadeias alfa no citoplasma da hemácia. As cadeias alfa livres são completamente insolúveis e precipitam no citoplasma da célula. O seu efeito tóxico culmina na destruição dos eritroblastos na própria medula óssea, um processo chamado eritropoiese ineficaz. Na beta-talassemia maior, somente 15-30% dos eritroblastos escapam da destruição medular.
As pequenas células sobreviventes originam hemácias defeituosas, além de microcíticas e hipocrômicas, apresentam alguns corpúsculos de inclusão remanescentes da cadeia alfa, que as tornam suscetíveis aos macrófagos do baço, explicando a hemólise extravascular crônica.
Ainda há um outro problema… A eritropoiese ineficaz acaba estimulando (por um mecanismo desconhecido) a absorção intestinal de ferro, mesmo na ausência da reposição inadvertida de sulfato ferroso, podendo levar a hemossiderose (ou hemocromatose).
Os Achados Laboratoriais
Os principais achados laboratoriais são de uma anemia grave, microcítica e hipocrômica, com VCM entre 48-72 fl e CHCM entre 23-32 g/dl. As alterações características de hemólise extravascular encontram-se presentes: hiperbilirrubinemia indireta, aumento do LDH, redução da haptoglobina, reticulocitose.
Como a anemia não é exclusivamente pela hemólise, o índice de reticulócitos é de apenas 5-15%, menor do que o esperado para a gravidade da anemia. O hemograma pode apresentar leucocitose neutrofílica, mas com plaquetas normais. O esfregaço do sangue periférico é bastante rico. Há uma intensa anisocitose e poiquilocitose, com predomínio de hemácias microcíticas e hipocrômicas e de hemácias em alvo.
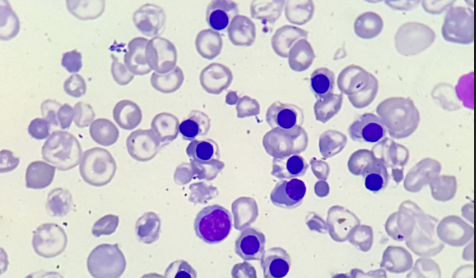
Os eritroblastos na periferia (“hemácias nucleadas”), correspondem a mais de 10% da contagem leucocitária, ou mais de 200/mm³. Eventualmente, pode aparecer também hemácias com pontilhados basofílicos tal como na anemia sideroblástica.

Referências
ZAGO M. A. TRATADO DE HEMATOLOGIA São Paulo: Editora Atheneu, 2013.
LORENZI T. F. ATLAS DE HEMATOLOGIA: Clínica Hematológica Ilustrada. Edição 2006 Rio de Janeiro:Guanabara Koogan
