A hemoglobinopatia C é uma condição genética rara que altera a estrutura da hemoglobina. Essa alteração é causada por uma mutação específica no gene da cadeia beta da hemoglobina, onde o aminoácido glutâmico é substituído por lisina na posição 6 da cadeia beta.
Essa mudança resulta na produção de hemoglobina C, que pode formar cristais dentro dos glóbulos vermelhos em determinadas condições, prejudicando sua função normal de transporte de oxigênio. Existem três haplótipos descritos: CI, CII e CIII. A frequência desses polimorfismos sugere uma possível vantagem seletiva para os portadores heterozigotos, como uma menor parasitemia por Plasmodium falciparum, embora não tão evidente quanto a conferida pela hemoglobina S.
A hemoglobina C altera a troca de íons pela membrana das hemácias e modifica sua forma, resultando em células em alvo, microcitose, hipocromia e esferócitos no esfregaço sanguíneo. A hemoglobina anormal promove a perda de potássio nas células vermelhas, diminuindo sua vida média, que ainda é superior à dos eritrócitos com hemoglobina S.
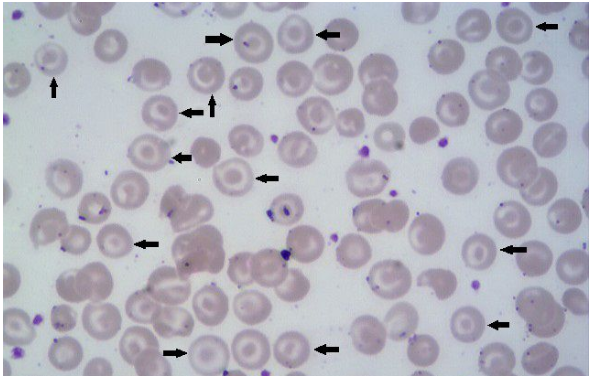
As manifestações clínicas e laboratoriais significativas geralmente ocorrem em indivíduos com herança homozigótica (CC) ou em associação com a HbS (SC) e talassemias. Portadores do traço C (HbC < 50%) são assintomáticos e não apresentam anemia ou alteração do VCM, embora possam ser observadas hemácias em alvo no esfregaço sanguíneo. Em contraste, os homozigotos (CC) apresentam anemia microcítica de intensidade leve, esplenomegalia moderada e frequentes hemácias em alvo, além de ocasionalmente cristais de HbC. Na associação entre HbC e talassemia beta (HbCF), ocorre anemia microcítica e hipocrômica de grau discreto.
A hemoglobina C possui uma afinidade ligeiramente reduzida pelo oxigênio. As variações de volume devido a alterações na pressão osmótica são sutis, e a lise osmótica ocorre em valores de VCM menores do que nas células normais com hemoglobina A (Hb A). Isso se deve à redução da fragilidade osmótica, resultante do aumento da relação superfície-volume celular, uma vez que a célula se desidrata e a concentração de hemoglobina corpuscular média (CHCM) se eleva.
Embora menos comum que outras condições, como a doença falciforme, a hemoglobinopatia C apresenta desafios clínicos significativos, especialmente em regiões onde é mais prevalente. O diagnóstico e a gestão adequados são essenciais para prevenir complicações e melhorar a qualidade de vida dos afetados. A conscientização e o rastreamento genético são importantes para identificar e tratar essa condição, especialmente em populações de risco.

Referências
ANGULO, Ivan L.; PICADO, Sandra BR. Hemoglobina C em homozigose e interação com talassemia beta. Revista Brasileira de Hematologia e Hemoterapia, 2009.
Naoum, Flávio Augusto. Doenças que alteram os exames hematológicos / Flávio Augusto Naoum. – 2. ed. – Rio de Janeiro: Atheneu, 2017
