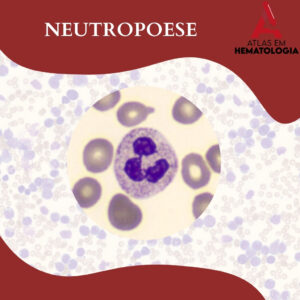
NEUTROPOESE: IDENTIFICANDO A MORFOLOGIA DA LINHAGEM NEUTROFÍLICA
Leitura: 4 min
A neutropoese é o processo hematopoiético responsável pela produção e maturação dos neutrófilos, que são leucócitos essenciais na defesa contra infecções bacterianas e fúngicas principalmente. A linhagem neutrofílica, originada na medula óssea, segue uma série […]