TALASSEMIAS
Classificação e Alterações Morfológicas
As talassemias são um grupo de doenças hereditárias caracterizadas pela produção defeituosa de hemoglobina, levando a anemia de gravidade variável. A hemoglobina é composta por cadeias de globina (alfa e beta), e a disfunção na produção dessas cadeias leva a dois tipos principais de talassemia: beta-talassemia e alfa-talassemia. A alteração estrutural e disfunção da hemoglobina nessa doença resultam em destruição excessiva dos glóbulos vermelhos , o que leva à anemia.
Nas talassemias, há uma anormalidade de um ou mais genes da globina, levando à redução da produção de proteína globina. Há, portanto, um desequilíbrio da síntese da cadeia de globina beta e alfa, levando à precipitação da cadeia de globina , eritropoiese ineficaz e hemólise .
Quase todo o oxigênio transportado pelo sangue está ligado à hemoglobina. Ao passarem pelos pulmões, os eritrócitos têm suas moléculas de hemoglobina saturadas em 96% de oxigênio (oxi hemoglobina do sangue arterial), que será gradualmente liberado para os tecidos. No sangue venoso, que retorna ao coração, a hemoglobina está apenas 64% saturada de oxigênio. Assim, o sangue que passa através dos tecidos libera cerca de um terço do oxigênio que transporta. A molécula de hemoglobina é um tetrâmero constituído por 4 cadeias polipeptídicas, conhecidas como globinas, que geralmente são:
- 2 cadeias alfa , cada uma com 141 aminoácidos de comprimento
- 2 cadeias beta , cada uma com 146 aminoácidos de comprimento
Ligada a cada cadeia está uma molécula contendo ferro conhecida como heme. O oxigênio é transportado em combinação com a molécula de ferro do grupo heme (esta é uma reação de oxigenação, não de oxidação).
Fisiopatologia e Classificação das Talassemias
Na talassemia, a redução na síntese de cadeias de globina causa um desequilíbrio entre as cadeias de alfa e beta globina. Esse desequilíbrio leva à precipitação de cadeias de globina não pareadas nas hemácias, resultando em lesão celular e destruição prematura das hemácias (hemólise).
Talassemia Beta
Essa condição resulta de uma redução total (β⁰) ou parcial (β⁺) na produção de globinas beta, levando ao acúmulo de globinas alfa nos precursores eritroides. Na forma heterozigota (talassemia beta menor), os portadores apresentam uma condição geralmente assintomática, com anemia leve e alterações morfológicas como microcitose e hipocromia.
A forma homozigota, chamada talassemia beta maior, causa anemia grave desde a infância, provocando deformidades ósseas, atraso no crescimento e a necessidade de transfusões de sangue regulares, o que pode levar à sobrecarga de ferro e, sem tratamento adequado, a danos no coração e fígado. A talassemia intermediária abrange um espectro clínico variável, entre uma condição mais grave que a talassemia menor e uma forma próxima à talassemia maior.
Na eletroforese, a talassemia beta menor é marcada pelo aumento de HbA₂, enquanto na talassemia beta maior há uma diminuição ou ausência de HbA, com aumento de HbF. Na forma intermediária, os níveis de HbF variam conforme a gravidade da mutação.
Na ausência ou redução da síntese de cadeias beta, há um excesso de cadeias alfa livres, que são instáveis e formam agregados tóxicos dentro dos precursores eritroides na medula óssea. Esses agregados causam morte celular precoce (eritropoiese ineficaz) e, nas hemácias que sobrevivem e são liberadas para a circulação, há uma maior propensão à hemólise no baço, levando à anemia crônica.
Talassemia Alfa
A talassemia alfa é a doença monogênica mais comum no mundo, essa condição é causada pela deleção total (α⁰) ou parcial (α⁺) de um ou mais genes alfa, comprometendo a produção de globinas alfa e gerando um desequilíbrio com outras globinas que continuam sendo produzidas normalmente.
Durante o período fetal, o excesso de globinas gama forma a hemoglobina Bart’s. Após o sexto mês de vida, o excesso de globinas beta leva à formação de hemoglobina H, resultando em anemia hemolítica. A alteração nos índices hematimétricos se torna evidente apenas quando dois genes alfa são afetados, mas a presença de HbH pode ser detectada por eletroforese.
A forma mais grave, causada pela deleção de três genes, gera anemia microcítica e hipocrômica moderada, com esplenomegalia e necessidade ocasional de transfusões. A deleção de quatro genes provoca hidropsia fetal, condição fatal no útero ou logo após o nascimento. O diagnóstico é confirmado pela detecção de Hb Bart’s ou HbH por eletroforese e outros exames laboratoriais.
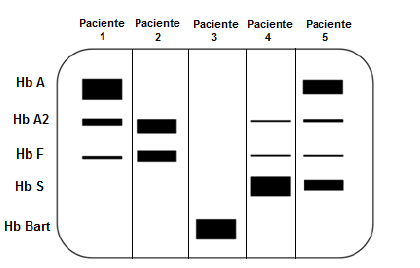
Padrão de Eletroforese de hemoglobinas onde: Paciente 1: adulto normal; Paciente 2: quadro de Beta talassemia heterozigótica – minor (traço talassêmico); Paciente 3: quadro de Alfa talassemia; Paciente 4: quadro de anemia falciforme homozigótica; Paciente 5: quadro de anemia falciforme heterozigótica.
Alterações Morfológicas
As talassemias são tipicamente associadas a alterações morfológicas das hemácias, que podem ser observadas em esfregaços de sangue periférico. As características incluem:
- Microcitose: As hemácias são menores que o normal devido à redução da síntese de hemoglobina.
- Hipocromia: As células têm menos cor devido à menor quantidade de hemoglobina.
- Anisocitose: Variação no tamanho das hemácias.
- Poiquilocitose: Variação na forma das hemácias, frequentemente observando-se hemácias em alvo (células em alvo ou codócitos), que são células com um acúmulo central de hemoglobina.
- Hemácias fragmentadas: Devido à destruição prematura.
- Corpos de Heinz: Presença de agregados de hemoglobina desnaturada nas hemácias (particularmente na alfa-talassemia).
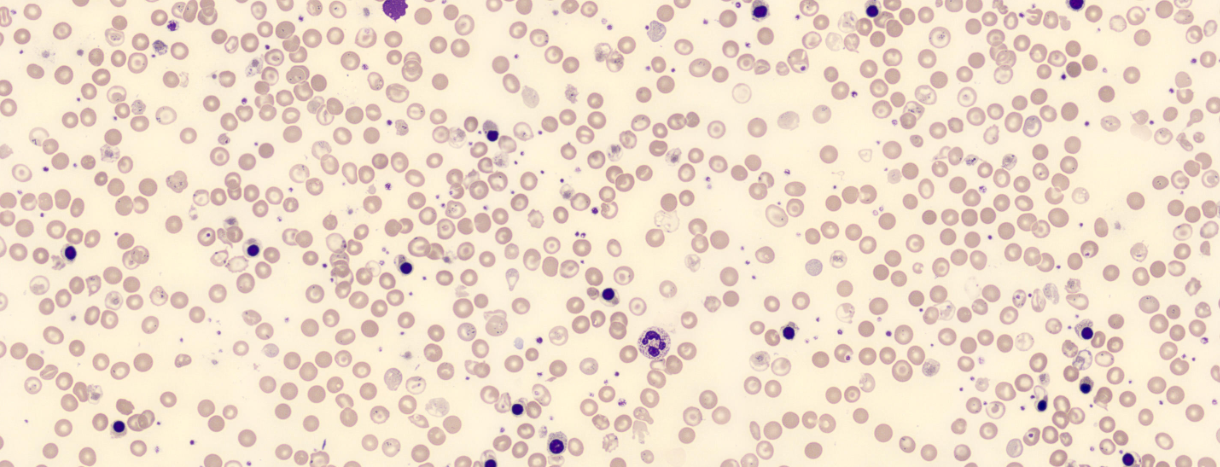
Lâmina de paciente com talassemia, com presença de hipocromia e poliquilocitose com presença intensa de codócitos, além de eritroblastos devido intensa hemólise e hiperatividade medular. Fonte: Cell\wiki
Na beta-talassemia maior, a medula óssea expande-se para tentar compensar a anemia, resultando em deformidades ósseas. Além disso, o baço e o fígado podem aumentar de tamanho devido ao aumento da hemólise e da eritropoiese extramedular.
É importante ressaltar que a fisiopatologia das formas leves, intermediárias e graves das talassemias, notadamente as do tipo beta, está pautada no fenômeno da eritropoiese ineficaz, em que o principal componente responsável pela anemia dos pacientes é a hemólise intramedular, ou seja, a destruição dos precursores eritroides mal formados dentro da medula óssea. De menor importância fisiopatológica em relação à intensidade da anemia, encontra-se a parcela de hemólise que ocorre na periferia, principalmente no baço.
Em conclusão, as talassemias são distúrbios genéticos graves, principalmente nas suas formas maiores, como a beta-talassemia maior e a doença da hemoglobina H. A fisiopatologia envolve desequilíbrios na produção de cadeias de globina, levando à hemólise e eritropoiese ineficaz. As alterações morfológicas das hemácias são características diagnósticas importantes e refletem a gravidade da anemia e o tipo de talassemia. O tratamento, especialmente nas formas mais graves, pode envolver transfusões regulares, terapia quelante de ferro e, em casos selecionados, transplante de medula óssea.
O próximo passo de todo analista que deseja ter mais segurança na bancada
Todo analista que busca se destacar e se tornar um profissional mais atualizado, capacitado e qualificado para o mercado de trabalho precisa considerar uma pós-graduação.
Um profissional com especialização é valorizado na área laboratorial; esse é um fato inegável.
Unimos o útil ao agradável ao desenvolver uma pós-graduação em Hematologia Laboratorial e Clínica.
Para aqueles que procuram a comodidade de uma pós-graduação 100% online e ao vivo, sem abrir mão da excelência no ensino, temos a solução ideal.
Nossa metodologia combina teoria e prática da rotina laboratorial, garantindo um aprendizado efetivo.
Contamos com um corpo docente altamente qualificado, com os melhores professores do Brasil, referências em suas áreas de atuação.
No Instituto Nacional de Medicina Laboratorial, temos apenas um objetivo: mais do que ensinar, vamos tornar VOCÊ uma referência.
Toque no botão abaixo e conheça a pós-graduação em Hematologia Laboratorial e Clínica.
QUERO CONHECER TODOS OS DETALHES DA PÓS-GRADUAÇÃO
Referências
Naoum, Flávio Augusto. Doenças que alteram os exames hematológicos. 2. ed. Rio de Janeiro: Atheneu, 2017.
MELGAREJO, Celsa Raquel Villaverde. Beta talassemia menor: aspectos clínicos e laboratoriais. 2015.
PIGNATTI, Caterina Borgna; GALANELLO, Renzo. Talassemia e Distúrbios Relacionados: Distúrbio Quantitativo da Síntese de Hemoglobina. Wintrobe’s Clinical Haematology. Filadélfia: Lippincott Williams & Wilkins, v. 1084, 2009.


0 Comentários