ANEMIA APLÁSTICA
Fisiopatologia e Alterações na Hematopoiese
A anemia aplástica é uma condição rara caracterizada pela pancitopenia, ou seja, uma redução simultânea no número de eritrócitos, leucócitos e plaquetas, causada pela falência da medula óssea em produzir células hematopoiéticas. A prevalência é de 2 a 5 casos por milhão de habitantes por ano, sendo mais comum em países asiáticos. Ess distúrbio pode ser classificado como congênita ou adquirida, sendo a forma adquirida a mais prevalente.
A fisiopatologia da anemia aplástica envolve a destruição ou incapacidade das células-tronco hematopoiéticas da medula óssea de se replicarem e se diferenciarem de forma adequada. Os mecanismos envolvidos podem ser autoimunes, tóxicos, infecciosos ou genéticos, resultando na falência da medula óssea em produzir as células sanguíneas.
Aplasia Primária (idiopática) da Medula Óssea
A aplasia primária é caracterizada pela presença de pancitopenia no sangue periférico e hipoplasia da medula óssea. Esse tipo de aplasia representa aproximadamente metade dos casos de anemia aplástica adquirida, nos quais não se consegue identificar uma causa subjacente. O mecanismo exato ainda não está completamente esclarecido, mas acredita-se que uma reação autoimune contra as células precursoras hematopoiéticas possa estar envolvida.
Clinicamente, os pacientes apresentam fraqueza, cansaço, infecções frequentes e tendência a sangramentos. No hemograma, há pancitopenia de intensidade variável, sem alterações morfológicas significativas. A anemia pode ser normocítica ou macrocítica, enquanto a leucopenia é dominada pela neutropenia. A biópsia de medula óssea geralmente confirma a hipocelularidade, com celularidade inferior a 25%.
Aplasia Secundária da Medula Óssea
A aplasia secundária está associada a fatores subjacentes identificáveis, como infecções virais (hepatite C, HIV), exposição à radiação, certas drogas (como cloranfenicol, antimaláricos, anti reumáticos) e quimioterápicos. No hemograma, pode-se observar citopenia em uma ou mais linhagens celulares, e a medula óssea se apresenta hipocelular ou com sinais de displasia. O tratamento da causa subjacente pode levar à melhora do quadro hematológico.
Aplasias Congênitas
As aplasias congênitas são síndromes hereditárias raras, caracterizadas por anormalidades nos precursores hematopoiéticos e anemia aplástica, manifestando-se na infância ou adolescência. A mais comum é a anemia de Fanconi, que apresenta herança autossômica recessiva ou ligada ao cromossomo X e afeta genes responsáveis pelos mecanismos de reparo do DNA, provocando instabilidade cromossômica. Além de macrocitose, neutropenia e plaquetopenia no hemograma, os pacientes podem apresentar alterações cutâneas, esqueléticas, urogenitais e risco elevado de malignidades. Outra aplasia congênita é a disceratose congênita, relacionada a defeitos nas enzimas que mantêm o comprimento dos telômeros, como a telomerase, manifestando-se tardiamente com distrofias ungueais e leucoplasias. A anemia de Blackfan-Diamond, caracterizada por aplasia eritroide pura, pode também evoluir para neutropenia e plaquetopenia.
A principal alteração morfológica na anemia aplástica ocorre na medula óssea, onde se observa uma drástica redução da celularidade, com substituição das células hematopoiéticas por gordura e estroma. Normalmente, a medula saudável contém entre 30% e 70% de células hematopoiéticas, dependendo da idade. Na anemia aplástica, essa porcentagem cai para menos de 25%. No hemograma, é comum observar pancitopenia, com anemia, e baixa contagem de reticulócitos.
A leucopenia, especialmente neutropenia, eleva o risco de infecções graves, enquanto a trombocitopenia predispõe a sangramentos, visíveis como petéquias e equimoses. O exame de esfregaço periférico revela pouca ou nenhuma alteração morfológica significativa nos eritrócitos, que podem apresentar formas ligeiramente irregulares, mas sem as características marcantes de outras anemias.
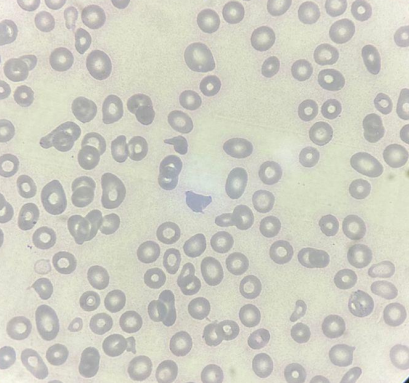
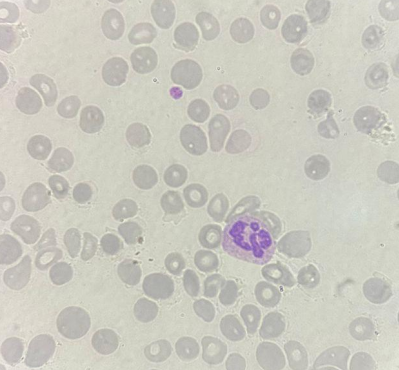
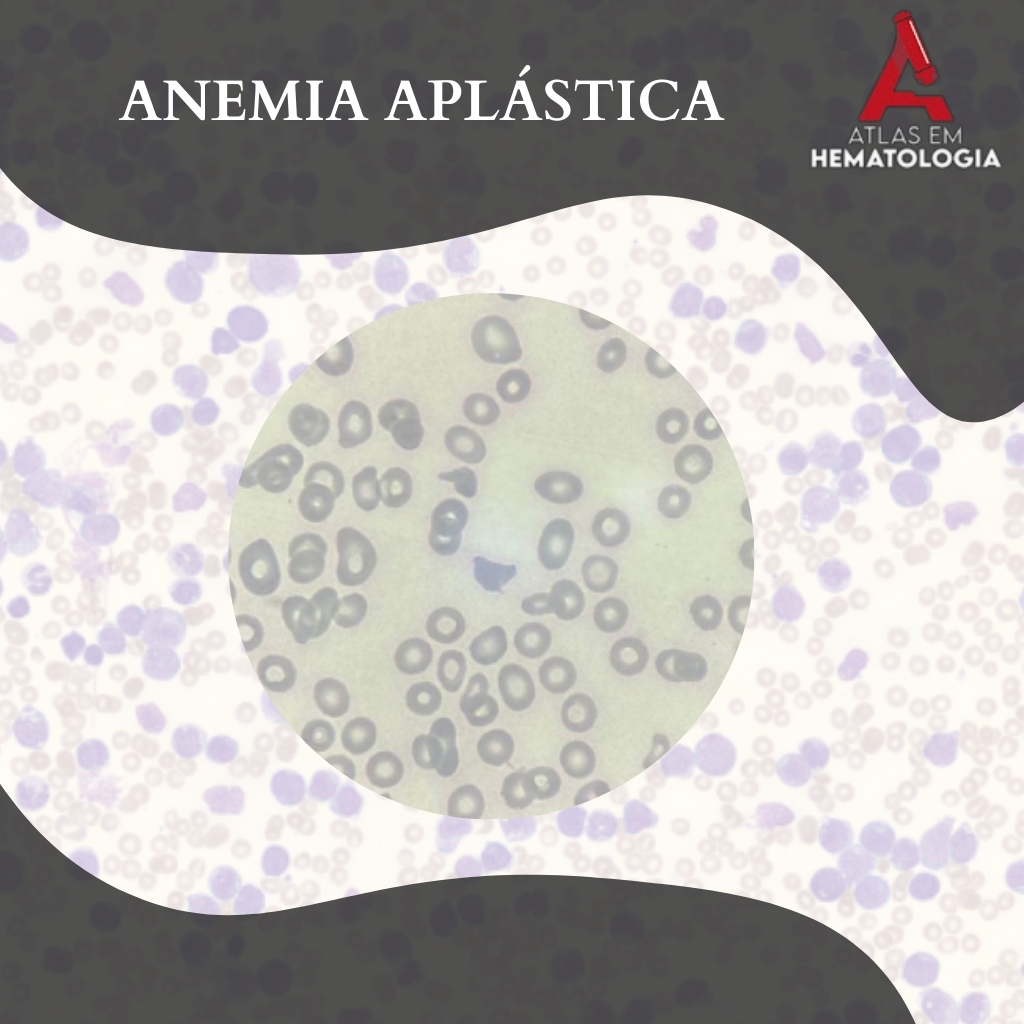
Lâmina de aplasia com hipocromia, policromasia e baixa celularidade. Fonte: CellWiki
Em resumo, a anemia aplástica é uma desordem hematológica rara, caracterizada pela falência da medula óssea em produzir células sanguíneas suficientes, resultando em pancitopenia. Embora a forma adquirida seja mais comum, fatores congênitos também desempenham um papel significativo em casos hereditários. A fisiopatologia envolve uma variedade de mecanismos, incluindo processos autoimunes, tóxicos, infecciosos e genéticos, com a medula óssea apresentando hipocelularidade como principal achado morfológico.
O diagnóstico depende do hemograma, mielograma e biópsia de medula óssea, que revelam as características morfológicas típicas da condição. A compreensão dos mecanismos subjacentes e das alterações morfológicas é essencial para o manejo adequado da anemia aplástica, com enfoque no tratamento das causas subjacentes e suporte hematológico, visando melhorar o prognóstico dos pacientes e a sua qualidade de vida.
O próximo passo de todo analista que deseja ter mais segurança na bancada
Todo analista que busca se destacar e se tornar um profissional mais atualizado, capacitado e qualificado para o mercado de trabalho precisa considerar uma pós-graduação.
Um profissional com especialização é valorizado na área laboratorial; esse é um fato inegável.
Unimos o útil ao agradável ao desenvolver uma pós-graduação em Hematologia Laboratorial e Clínica.
Para aqueles que procuram a comodidade de uma pós-graduação 100% online e ao vivo, sem abrir mão da excelência no ensino, temos a solução ideal.
Nossa metodologia combina teoria e prática da rotina laboratorial, garantindo um aprendizado efetivo.
Contamos com um corpo docente altamente qualificado, com os melhores professores do Brasil, referências em suas áreas de atuação.
No Instituto Nacional de Medicina Laboratorial, temos apenas um objetivo: mais do que ensinar, vamos tornar VOCÊ uma referência.
Toque no botão abaixo e conheça a pós-graduação em Hematologia Laboratorial e Clínica.
QUERO CONHECER TODOS OS DETALHES DA PÓS-GRADUAÇÃO
Referências
Naoum, Flávio Augusto. Doenças que alteram os exames hematológicos. 2. ed. Rio de Janeiro: Atheneu, 2017.
CAMPOS, R. X. et al. Anemia aplástica secundária à doença de Behçet: relato de caso e revisão da literatura. Hematology, Transfusion and Cell Therapy, v. 44, p. S53, 2022.
VAZ, Catarina Cruz. Anemia Aplástica Adquirida. 2016.




0 Comentários