DOENÇA DE VON WILLEBRAND
A doença de von Willebrand (DvW) é a desordem hemorrágica hereditária mais comum no mundo, afetando aproximadamente 1% da população geral. Caracterizada pela deficiência ou disfunção do fator de von Willebrand (FvW), essa condição compromete a hemostasia, resultando em episódios hemorrágicos que variam de leves a graves. A DvW está associada a uma série de manifestações clínicas, o que a torna um desafio diagnóstico e terapêutico.
Estrutura e Função do FvW
O FvW é uma glicoproteína grande e multimérica, secretada pelo endotélio e pelos megacariócitos, que apresenta duas funções básicas: promover a adesão plaquetária e carrear e estabilizar o fator VIII da coagulação.
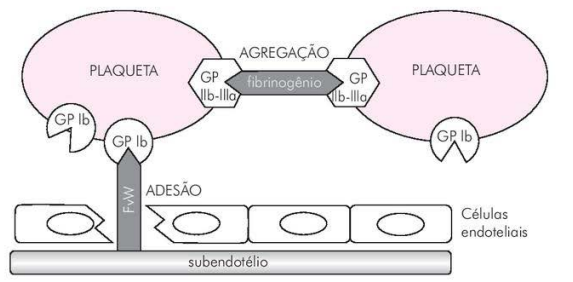
Adesão e agregação plaquetária. O FvW promove a ligação entre a glicoproteína Ib/IX (GP Ib) e o colágeno do subendotélio (adesão plaquetária).Fonte: Academia de Clínica Hematológica (AC&H).
É produzido principalmente por células endoteliais e megacariócitos. A proteína existe em diferentes formas, variando em tamanho e função:
- Multímeros: O FvW circula no plasma em multímeros, que são longas cadeias de polipeptídeos. Essa estrutura multimérica permite que o FvW tenha uma grande área de superfície, essencial para suas funções biológicas como a adesão plaquetária, especialmente em lesões vasculares
- Interação com Plaquetas: O FvW se liga ao colágeno exposto durante a lesão vascular, facilitando a adesão plaquetária. A ligação do vWF ao receptor glicoproteína Ib/IX/V nas plaquetas é essencial para a formação do tampão hemostático.
- Estabilização do Fator VIII: O vWF também atua como transportador do fator VIII, protegendo-o da degradação pelo sistema do complemento. A redução do FvW pode levar à diminuição da atividade do fator VIII, comprometendo a cascata de coagulação.
Tipos de Doença de von Willebrand
A Doença de von Willebrand (DvW) é classificada em três tipos principais, cada um apresentando características clínicas e laboratoriais distintas:
Tipo 1: O Tipo 1 da DvW é caracterizado por uma deficiência leve a moderada do fator de von Willebrand (vWF). Neste tipo, os níveis de FvW e do fator VIII são geralmente normais, mas a quantidade total de FvW está reduzida. Essa redução pode ser suficiente para causar manifestações hemorrágicas leves a moderadas, como epistaxe (sangramento nasal), hematomas frequentes e, em alguns casos, sangramentos gengivais. Os pacientes com o Tipo 1 geralmente não apresentam sintomas severos, a condição pode ter as suspeitas iniciais através de um hemograma (plaquetas em contagem normal) e testes de função plaquetária (alterado).
Tipo 2: O Tipo 2 da DvW é caracterizado por uma deficiência qualitativa do FvW e é subdividido em quatro subtipos: 2A, 2B, 2M e 2N.
- Tipo 2A: Este subtipo é marcado pela dificuldade na formação de multímeros grandes do FvW, resultando em adesão plaquetária inadequada. A ausência de multímeros grandes compromete a função plaquetária, levando a episódios hemorrágicos, como hematomas e sangramentos prolongados após lesões ou procedimentos cirúrgicos.
- Tipo 2B: Neste subtipo, ocorre uma hipersensibilidade do FvW que resulta em uma adesão plaquetária excessiva. podendo levar à trombocitose (aumento do número de plaquetas) e ao consumo de plaquetas, resultando em plaquetopenia (diminuição do número de plaquetas). Os pacientes com Tipo 2B podem apresentar manifestações hemorrágicas semelhantes às do Tipo 2A, mas com características adicionais devido à trombocitose.
- Tipo 2M: O subtipo 2M é caracterizado por uma disfunção na ligação do vWF ao colágeno, sem alterações significativas na formação de multímeros. Embora os níveis totais de vWF possam estar normais, a função de adesão é comprometida, resultando em hemorragias que podem ser moderadas.
- Tipo 2N: O subtipo 2N apresenta uma afinidade reduzida do vWF pelo fator VIII, o que resulta em níveis diminuídos do fator VIII e, consequentemente, em hemorragias significativas devido a deficiência deste fator. Esse subtipo pode ser confundido com a hemofilia A, pois os pacientes apresentam sangramentos graves, mas o tratamento é diferente, enfatizando a importância de um diagnóstico preciso.
Tipo 3: O Tipo 3 da DvW é a forma mais severa da doença, caracterizada por uma deficiência quase total ou indetectável do vWF. Os pacientes com Tipo 3 frequentemente apresentam hemorragias graves e complicações que podem incluir sangramentos espontâneos e hemorragias após traumas ou cirurgias, levando a um risco aumentado de hemorragias intracranianas e outros eventos hemorrágicos potencialmente fatais. Devido à gravidade da condição, esses pacientes frequentemente requerem intervenções mais agressivas, como transfusões de concentrados de vWF e fator VIII, especialmente em situações de emergência.
A avaliação laboratorial geral da doença de von Willebrand geralmente mostra plaquetas em níveis normais, com um tempo de sangramento prolongado. Em alguns casos, a diminuição da atividade do fator VIII pode levar a alterações no TTPA, enquanto o TP permanece normal. O teste de agregação plaquetária indica hipoagregação na presença de ristocetina, já que sua ação agonista depende do fator de von Willebrand (vWF). O tipo 2B é uma exceção, pois costuma apresentar plaquetopenia leve e agregação normal ou até aumentada com ristocetina. O diagnóstico específico geralmente requer a quantificação do antígeno do vWF, a análise do cofator da ristocetina, a dosagem do fator VIII e, quando disponível, a avaliação dos multímeros do vWF, permitindo assim a identificação do subtipo.
Assim, o entendimento aprofundado da fisiopatologia do vWF, sua interação com o sistema plaquetário e os mecanismos subjacentes à hemorragia são essenciais para o manejo eficaz dos pacientes. Com os avanços na terapia e no diagnóstico, a qualidade de vida dos indivíduos afetados pode ser significativamente melhorada. A pesquisa contínua é crucial para descobrir novas terapias e abordagens que possam transformar o cuidado desses pacientes, promovendo um futuro com menos complicações hemorrágicas.
O próximo passo de todo analista que deseja ter mais segurança na bancada
Todo analista que busca se destacar e se tornar um profissional mais atualizado, capacitado e qualificado para o mercado de trabalho precisa considerar uma pós-graduação.
Um profissional com especialização é valorizado na área laboratorial; esse é um fato inegável.
Unimos o útil ao agradável ao desenvolver uma pós-graduação em Hematologia Laboratorial e Clínica.
Para aqueles que procuram a comodidade de uma pós-graduação 100% online e ao vivo, sem abrir mão da excelência no ensino, temos a solução ideal.
Nossa metodologia combina teoria e prática da rotina laboratorial, garantindo um aprendizado efetivo.
Contamos com um corpo docente altamente qualificado, com os melhores professores do Brasil, referências em suas áreas de atuação.
No Instituto Nacional de Medicina Laboratorial, temos apenas um objetivo: mais do que ensinar, vamos tornar VOCÊ uma referência.
Toque no botão abaixo e conheça a pós-graduação em Hematologia Laboratorial e Clínica.
QUERO CONHECER TODOS OS DETALHES DA PÓS-GRADUAÇÃO
Referências
REZENDE, Bruna Manzutti; FIGUEIREDO, Andréa Mendes. Diagnóstico laboratorial da Doença de Von Willebrand: uma revisão de literatura. SALUSVITA, v. 40, n. 2, p. 123-135, 2021.
Naoum, Flávio Augusto. Doenças que alteram os exames hematológicos. 2. ed. Rio de Janeiro: Atheneu, 2017.
JOÃO, Cristina. Doença de von Willebrand. Medicina Interna, v. 8, n. 1, p. 28-36, 2001.




0 Comentários